Scientists at the University of Pennsylvania Perelman School of Medicine have developed a new genetic approach to specifically block the damaged copy of the gene for a rare bone disease, while leaving the normal copy untouched (Gene Therapy, October 20, 2011).
Fibrodysplasia ossificans progressive (FOP) is a rare genetic disorder of progressive extra bone formation for which there is presently no cure. It is caused by a mutation in the gene for ACVR1/ALK2, a bone morphogenetic protein (BMP) receptor that occurs in all classically affected individuals. Individuals who have FOP harbor one normal copy and one damaged copy of the gene in each cell. The mutation increases the amount of BMP in cells to greater than normal levels, which initiates the transformation of muscles and cartilage into a disabling second skeleton of bone.
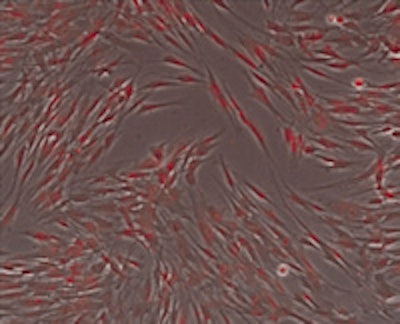
 Microscopic image of tooth-derived stem cells from an FOP patient acquired through the FOP Good Tooth Fairy Program at the Perelman School of Medicine at the University of Pennsylvania. Image courtesy of Josef Kaplan, PhD.
Microscopic image of tooth-derived stem cells from an FOP patient acquired through the FOP Good Tooth Fairy Program at the Perelman School of Medicine at the University of Pennsylvania. Image courtesy of Josef Kaplan, PhD.
Using a special type of RNA molecule engineered to specifically silence the damaged copy of the gene rather than the normal copy -- a process known as RNA interference -- the scientists restored the cellular function caused by the FOP mutation by ridding cells of the mutant ACVR1/ALK2 mRNA. Cells were essentially left with only normal copies of ACVR1/ALK2 mRNA, thus adjusting the protein's activity to normal, similar to that of cells without the FOP mutation.
The human cells used in the experiments were adult stem cells obtained directly from discarded baby teeth donated by FOP patients. These contained the exact combination of damaged and normal ACVR1/ALK2 receptor proteins found in all classically affected FOP patients worldwide. The discarded teeth were obtained from FOP pediatric patients and normal controls, usually nonaffected siblings, in the ongoing FOP Good Tooth Fairy Program.
The utility of this approach must be confirmed in mouse models of classic FOP prior to its consideration for human use, the authors cautioned. Additionally, other hurdles stand in the way of human application at the present time, most notably safe delivery of the small RNA molecules to cells in the human body.